1 How CRISPR-Cas9 works
CRISPR-Cas9 is a powerful gene-editing system that enables precise cleavage of double-stranded DNA at specific target sites.
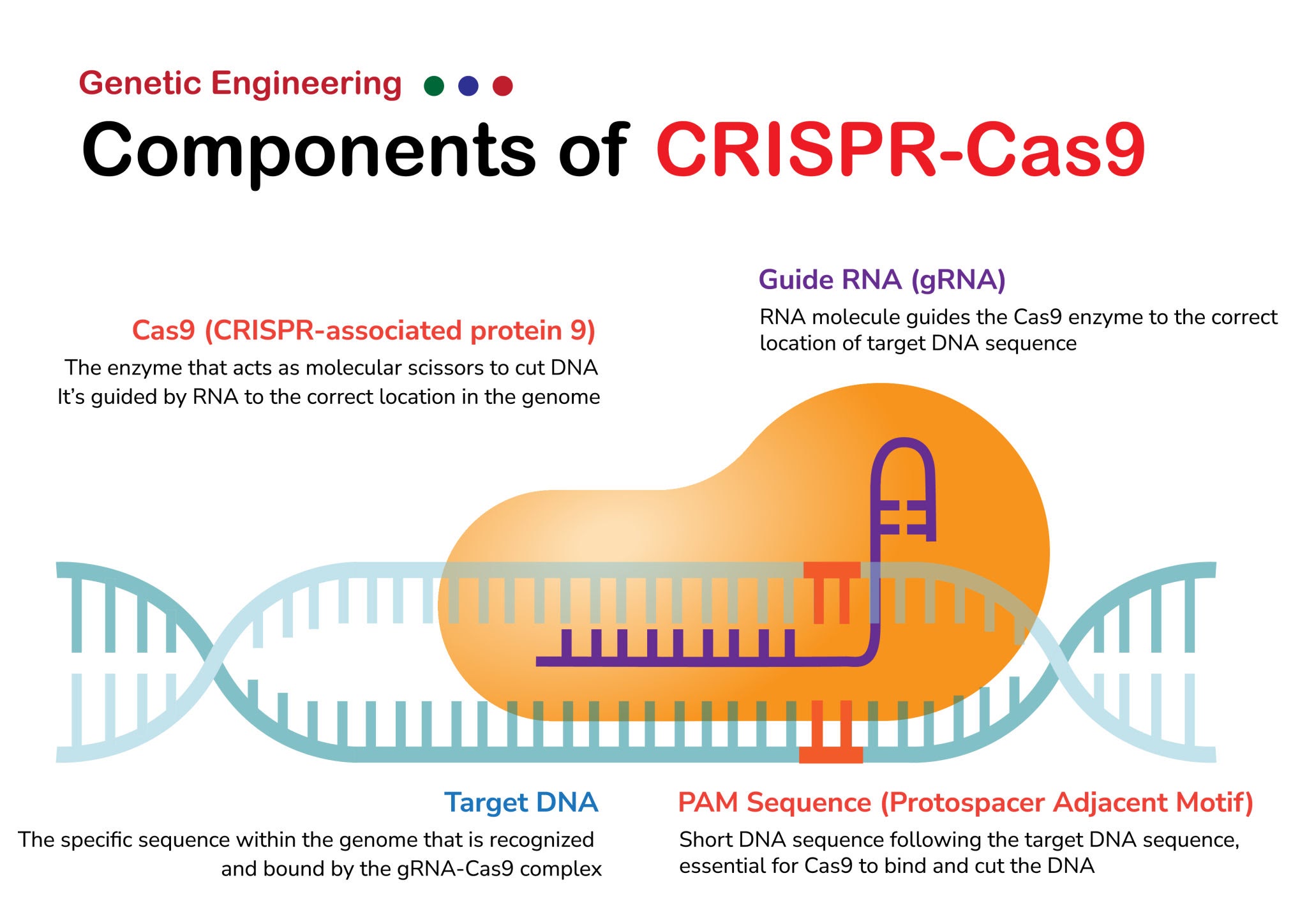
gRNA 的一端含有与目标 DNA 序列互补的 20 个核苷酸序列,即引导序列,它负责识别并引导 Cas9 到达基因组中的特定位置。
Cas9 酶:DNA 切割酶。
PAM,即间隔区相邻基序:只有在 gRNA-Cas9 complex 识别了 PAM 之后(对于 Cas9,其 PAM 序列通常是 NGG),gRNA 才能与目标 DNA 进行互补配对,并最终引发 Cas9 对 DNA 的切割。
换句话说,DNA 序列必须包含 NGG,而后 NGG 上游紧邻的 20 个核苷酸序列才能被用于设计 gRNA。
切割发生在 NGG 与 20 个核苷酸序列之间。
Cas9 酶造成双链断裂(Double-Stranded Break, DSB)后会激活细胞自身的 DNA 修复机制:
非同源末端连接(Non-Homologous End Joining):细胞直接将断裂的两个 DNA 末端连接起来,容易出现 indels。对于 coding sequence,容易造成 frameshift mutation,从而产生无功能的截短蛋白或不稳定的 mRNA,相当于敲除了目标基因。
同源重组修复(Homology-Directed Repair, HDR):细胞利用一个同源 DNA 模板来修复 DSB,这可以实现特定序列的敲入。
“同源”指的是引入的 DNA 模板与切割位点两侧的序列相同或高度相似。
同源 DNA 模板包含三个必要部分:
左同源臂(Left Homology Arm, LHA):一段 DNA 序列,与 Cas9 切割点上游的序列完全相同。
右同源臂(Right Homology Arm, RHA):一段 DNA 序列,与 Cas9 切割点的下游序列完全相同。
插入片段(Cargo):位于 LHA 和 RHA 之间,是想要敲入基因组的新序列。
For more info, see this paper.
2 How to deploy CRISPOR locally
For detailed info, see CRISPOR online analysis platform and CRISPOR GitHub repo.
# clone the repo
cd /home/yangrui/softwares
git clone https://github.com/maximilianh/crisporWebsite.git
# use a conda env to install dependencies
micromamba create -c conda-forge -n crispor_cmd_env python=3.9
micromamba activate crispor_cmd_env
micromamba install bioconda::bwa
micromamba install conda-forge::pip
micromamba install conda-forge::matplotlib
pip install --trusted-host mirrors.tuna.tsinghua.edu.cn -i https://mirrors.tuna.tsinghua.edu.cn/pypi/web/simple biopython numpy scikit-learn pandas twobitreader xlwt keras tensorflow h5py rs3 pytabix matplotlib lmdbm
# re-train the Azimuth model and save it again
pip install --trusted-host mirrors.tuna.tsinghua.edu.cn -i https://mirrors.tuna.tsinghua.edu.cn/pypi/web/simple openpyxl
cd /home/yangrui/softwares/crisporWebsite/bin/Azimuth-2.0
mkdir saved_models
python model_comparison.py
rm -rf azimuth/saved_models
mv saved_models azimuth
# test the script
python /home/yangrui/softwares/crisporWebsite/crispor.py --help
cd /home/yangrui/softwares/crisporWebsite
mv genomes.sample genomes
mkdir -p sampleFiles/mine
python /home/yangrui/softwares/crisporWebsite/crispor.py sacCer3 sampleFiles/in/sample.sacCer3.fa sampleFiles/mine/sample.sacCer3.tsv -o sampleFiles/mine/sample.sacCer3.mine.offs.tsv
# add genomes
# for detailed info, see https://crispor.gi.ucsc.edu/genomes/genomeInfo.all.tab
cd /data/database/data/raw/crisporWebsite/genomes
/usr/local/bin/Rscript /data/softwares/misc_r_scripts/download_crispor_genomes.R hg38 $(pwd)download_crispor_genomes.R:
#!/usr/local/bin/Rscript
# download_crispor_genomes.R
args <- commandArgs(trailingOnly = TRUE)
if (length(args) != 3) {
message("download_crispor_genomes.R <query_genome> <output_dir> <vcf|no_vcf>\n\nIf vcf given, download all files found; otherwise only download essential files.")
quit(save = "no", status = 1)
} else {
if (!(args[3] %in% c("vcf", "no_vcf"))) {
message("the 3rd argument is invalid, either 'vcf' or 'no_vcf'")
quit(save = "no", status = 1)
}
message("download ", args[1], " and save it in ", args[2], " with ", args[3])
}
suppressMessages(library(rvest))
suppressMessages(library(vroom))
suppressMessages(library(tidyverse))
suppressMessages(library(glue))
url_template <- "https://crispor.gi.ucsc.edu/genomes/{query_genome}/"
genome_info_file <- "https://crispor.gi.ucsc.edu/genomes/genomeInfo.all.tab"
# specify the query genome name here
query_genome <- args[1]
# the directory where you want to save files
work_dir <- args[2]
no_vcf <- ifelse(args[3] == "no_vcf", TRUE, FALSE)
message("pull ", genome_info_file, " and check the query genome name ...")
genome_info_df <- vroom(genome_info_file)
query_genome_info_df <- filter(genome_info_df, name == query_genome)
if (nrow(query_genome_info_df) == 0) {
stop("your query genome is not in ", genome_info_file)
} else if (nrow(query_genome_info_df) > 1) {
stop("your query genome matches too many items in ", genome_info_file)
}
message("the query genome name is valid")
output_dir <- file.path(work_dir, query_genome)
dir.create(output_dir, showWarnings = FALSE)
setwd(work_dir)
url_instance <- glue(url_template)
message("pull index.html from ", url_instance, " ...")
cmd_args <- c(
url_instance,
"-P", output_dir,
"-r", "-l1", "--no-parent", "-nd",
"--reject", "robots*"
)
output_msg <- system2(
command = "wget",
args = cmd_args,
wait = TRUE
)
message(paste0(output_msg, collapse = "\n"))
index_file <- file.path(output_dir, "index.html")
if (!file.exists(index_file)) {
stop("file ", index_file, " does not exist")
} else {
message("pull index.html (saved in ", index_file, ") successfully")
}
message("start to parsing ", index_file, " ...")
files <- read_html(index_file) %>%
html_elements("table") %>%
html_elements("td") %>%
html_elements("a") %>%
html_attr("href")
# filtering
files <- na.omit(files)
files <- trimws(files)
files <- files[files != ""]
files <- files %>%
str_subset("^/", negate = TRUE) %>%
str_subset("\\?C=", negate = TRUE) %>%
str_subset("^\\.\\.$", negate = TRUE) %>%
str_subset("/$", negate = TRUE)
if (no_vcf) {
files <- str_subset(files, paste0("^(", query_genome, "|genome)"))
}
file_links <- paste0(url_instance, files)
if (length(file_links) == 0) {
stop("parsing ", index_file, " failed")
} else {
message("\nthese files will be downloaded:\n\n", paste0(file_links, collapse = "\n"), "\n\n")
file_download_links_file <- file.path(output_dir, "file_download_links.txt")
message("parsing ", index_file, " successfully")
message("file links have been saved in ", file_download_links_file)
vroom_write_lines(file_links, file = file_download_links_file)
}
message("start to downloading files ...")
cmd_args <- c(
"-i", file_download_links_file,
"-P", output_dir,
"-nd"
)
output_msg <- system2(
command = "wget",
args = cmd_args,
wait = TRUE
)
message(paste0(output_msg, collapse = "\n"))
message("downloading all files done!")2.1 Examples to run CRISPOR in command line
#!/usr/bin/bash -e
uuid_str=$(</proc/sys/kernel/random/uuid)
work_dir=/data/tmp/yangrui_tmp
cd "${work_dir}"
vim "${uuid_str}".run_crispor.sh
chmod +x "${uuid_str}".run_crispor.sh
log_file="${uuid_str}".run_crispor.log
pid_file="${uuid_str}".run_crispor.pid
nohup time ./"${uuid_str}".run_crispor.sh &> "${log_file}" &
echo $! > "${pid_file}"
kill -0 "$(<"${pid_file}")" 2>/dev/null && echo running || echo finished
ps -p "$(<"${pid_file}")" >/dev/null && echo running || echo finished#!/usr/bin/bash -e
MAMBA_EXE=/home/yangrui/softwares/micromamba/bin/micromamba
CRISPOR_PATH=/home/yangrui/softwares/crisporWebsite/crispor.py
genome_dir=/data/database/data/raw/crisporWebsite/genomes
genome=mm10
# FASTA or BED file
input_file=2w_library.165bp.no_enzyme_cutting_sites.dealed.bed
pam=NGG
guide_out_file=${input_file}.${pam}.guides.tsv
off_target_file=${input_file}.${pam}.off_targets.tsv
# for speedup
# this line of code may cause some errors and is therefore commented out
# twoBitToFa ${genome_dir}/${genome}/${genome}.2bit /dev/shm/${genome}.fa
work_dir=$(pwd)
tmp_dir=${work_dir}/tmp_dir
mkdir ${tmp_dir}
${MAMBA_EXE} run -n crispor_cmd_env python ${CRISPOR_PATH} ${genome} ${input_file} ${guide_out_file} -o ${off_target_file} -p ${pam} -g ${genome_dir} --tempDir=${tmp_dir}过滤标准:
优先选取
GrafEtAlStatus == GrafOK:因为用于表达 gRNA 的启动子常为 Pol III 驱动的启动子。对于不是 GrafOK 的 gRNA,其末端 3-4 碱基包含特定的 T/C pattern,其和 Pol III 的转录终止信息十分相似,容易使 Pol III 从 DNA 模板上解离,从而停止转录。非 GrafOK 的 gRNA 会被 CRISPOR 标记为 Inefficient。优先选取 GC content <= 75% 的 gRNA:GC content > 75% 的会被 CRISPOR 标记为 High GC content。
cfdSpecScore越高越好。mitSpecScore越高越好。offtargetCount越低越好。